
Full text loading...
 , Brooke A. Schwartz1,2, Fantaysia C. Polanco2, Alexandra K. Firek3, Alayna C. Gumabong1, Nolan J. Hofstee1, Giri Narasimhan2,4, Trevor Cickovski2 and Kalai Mathee1,4
, Brooke A. Schwartz1,2, Fantaysia C. Polanco2, Alexandra K. Firek3, Alayna C. Gumabong1, Nolan J. Hofstee1, Giri Narasimhan2,4, Trevor Cickovski2 and Kalai Mathee1,4
Determining microbiome composition is a multi-step process. The 16S rRNA gene is amplified from the extracted DNA, sequenced, and then metagenomic data are input into a bioinformatic workflow. This paper describes how choices of bioinformatic pipelines, databases, and parameter values, such as maxEE and QTrim, can impact microbial compositional output.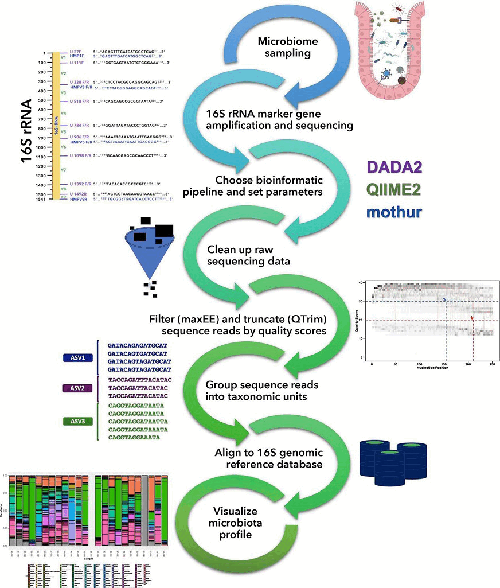
Introduction. The role of the microbiome in health and disease continues to be increasingly recognized. However, there is significant variability in the bioinformatic protocols for analysing genomic data. This, in part, has impeded the potential incorporation of microbiomics into the clinical setting and has challenged interstudy reproducibility. In microbial compositional analysis, there is a growing recognition for the need to move away from a one-size-fits-all approach to data processing.
Gap Statement. Few evidence-based recommendations exist for setting parameters of programs that infer microbiota community profiles despite these parameters significantly impacting the accuracy of taxonomic inference.
Aim. To compare three commonly used programs (DADA2, QIIME2, and mothur) and optimize them into four user-adapted pipelines for processing paired-end amplicon reads. We aim to increase the accuracy of compositional inference and help standardize microbiomic protocol.
Methods. Two key parameters were isolated across four pipelines: filtering sequence reads based on a whole-number error threshold (maxEE) and truncating read ends based on a quality score threshold (QTrim). Closeness of sample inference was then evaluated using a mock community of known composition.
Results. We observed that raw genomic data lost were proportionate to how stringently parameters were set. Exactly how much data were lost varied by pipeline. Accuracy of sample inference correlated with increased sequence read retention. Falsely detected taxa and unaccounted for microbial constituents were unique to pipeline and parameter. Implementation of optimized parameter values led to better approximation of the known mock community.
Conclusions. Microbial compositions generated based on the 16S rRNA marker gene should be interpreted with caution. To improve microbial community profiling, bioinformatic protocols must be user-adapted. Analysis should be performed with consideration for the select target amplicon, pipelines and parameters used, and taxa of interest.
Article metrics loading...
Full text loading...
References
Data & Media loading...
Supplements