

Full text loading...
Coinfections occurring within a single sample can be detected by exploring all mutations in a genomic set as these have full profiles commonly observed in lower prevalence.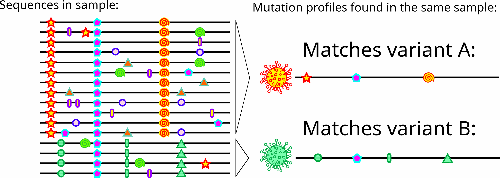
A total of 14 973 alleles in 29 661 sequenced samples collected between March 2021 and January 2023 by the Mexican Consortium for Genomic Surveillance (CoViGen-Mex) and collaborators were used to construct a thorough map of mutations of the Mexican SARS-CoV-2 genomic landscape containing Intra-Patient Minor Allelic Variants (IPMAVs), which are low-frequency alleles not ordinarily present in a genomic consensus sequence. This additional information proved critical in identifying putative coinfecting variants included alongside the most common variants, B.1.1.222, B.1.1.519, and variants of concern (VOCs) Alpha, Gamma, Delta, and Omicron. A total of 379 coinfection events were recorded in the dataset (a rate of 1.28 %), resulting in the first such catalogue in Mexico. The most common putative coinfections occurred during the spread of Delta or after the introduction of Omicron BA.2 and its descendants. Coinfections occurred constantly during periods of variant turnover when more than one variant shared the same niche and high infection rate was observed, which was dependent on the local variants and time. Coinfections might occur at a higher frequency than customarily reported, but they are often ignored as only the consensus sequence is reported for lineage identification.